Geometry optimization¶
Relax a molecular structure to its energy minimum. vibe-qc uses ASE’s
BFGS-line-search optimizer under the hood, driven by analytic HF/DFT
forces from the C++ core. run_job(..., optimize=True) is the
one-liner; the standard .out / .molden / .traj files all get
written as usual, plus the trajectory animation.
Water at HF/6-31G*¶
Starting from an approximate geometry:
from pathlib import Path
from vibeqc import Atom, Molecule, run_job
mol = Molecule([
Atom(8, [0.0, 0.0, 0.0 ]),
Atom(1, [0.0, 1.60, -1.10]), # slightly off-equilibrium
Atom(1, [0.0, -1.60, -1.10]),
])
run_job(
mol,
basis="6-31g*",
method="rhf",
output="water_opt",
optimize=True,
fmax=0.05, # eV/Å — converged when |F|_max drops below this
max_opt_steps=50,
)
Files produced:
water_opt.out— initial geometry, optimizer parameters, final optimized geometry, then the full SCF trace at the minimumwater_opt.molden— MOs at the minimum, for Avogadro / Jmolwater_opt.traj— one frame per BFGS step (ASE binary); view withase gui water_opt.traj
Reading the output¶
After the banner and atom table, water_opt.out contains:
Geometry optimisation (ASE/BFGS) -> water_opt.traj
fmax = 0.05 eV/A, max_steps = 50
Optimised geometry
Atoms (bohr)
------------------------------------------------------
1 Z= 8 0.00000000 0.00000000 0.07390216
2 Z= 1 0.00000000 1.44014211 -1.00364326
3 Z= 1 0.00000000 -1.44014211 -1.00364326
charge=0 multiplicity=1 n_electrons=10
iter energy (Ha) ...
(SCF trace at the optimised geometry)
...
converged in 7 iterations; E = -76.0108432849 Ha
The OH bond length and HOH angle at this HF/6-31G* minimum: r(OH) = 0.947 Å, HOH = 105.5° (experimental: 0.957 Å, 104.5°). HF slightly under-shoots the bond and opens the angle.
Parameters¶
Argument |
Default |
Meaning |
|---|---|---|
|
0.05 |
Force-convergence tolerance (eV/Å) |
|
200 |
Hard iteration limit for the optimizer |
|
|
Applied to every intermediate SCF; bump |
rhf_options / rks_options forward through to every frame of the
optimization, not just the final single point. This matters for
H-bonded systems and floppy molecules where one BFGS step sometimes
lands on a particularly stubborn geometry — bump max_iter = 200 or
add damping = 0.3 to the options struct and the whole trajectory
stays stable.
A harder case: the water dimer¶
H-bonded clusters sit on a very flat potential-energy surface — plain
BFGS Hessian extrapolation can push the waters apart before the
quadratic model is trustworthy. run_job uses BFGSLineSearch
which rejects uphill steps by construction, and pair this with a
realistic fmax:
from vibeqc import Atom, InitialGuess, Molecule, RHFOptions, run_job
mol = Molecule.from_xyz("h2o_dimer.xyz") # see examples/h2o_dimer.xyz
rhf_opts = RHFOptions()
rhf_opts.max_iter = 200 # H-bonded intermediates need it
rhf_opts.initial_guess = InitialGuess.SAD
run_job(
mol, basis="6-31g*", method="rhf",
output="water_dimer_opt",
optimize=True,
fmax=0.2, # force precision floor is ~0.1 eV/Å
max_opt_steps=25,
rhf_options=rhf_opts,
)
You’ll get R(OO) ≈ 2.98 Å (experimental: 2.976 Å) — the well-known
HF/6-31G* result. For a real binding-energy number add a correlation
method and dispersion; for a chemistry demonstration this is enough.
Visualizing the trajectory¶
ASE’s BFGS converges H₂O at HF/6-31G* in just 5 steps from a mildly distorted starting geometry. The trajectory below shows the molecule sliding into its equilibrium geometry while the energy drops by ~156 meV:
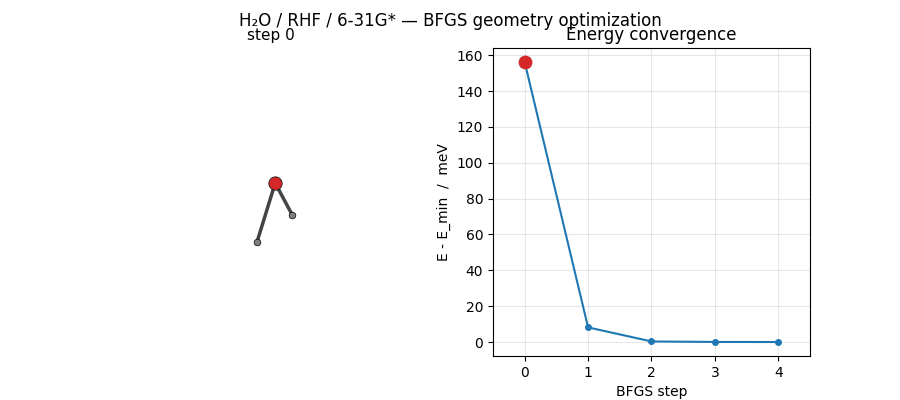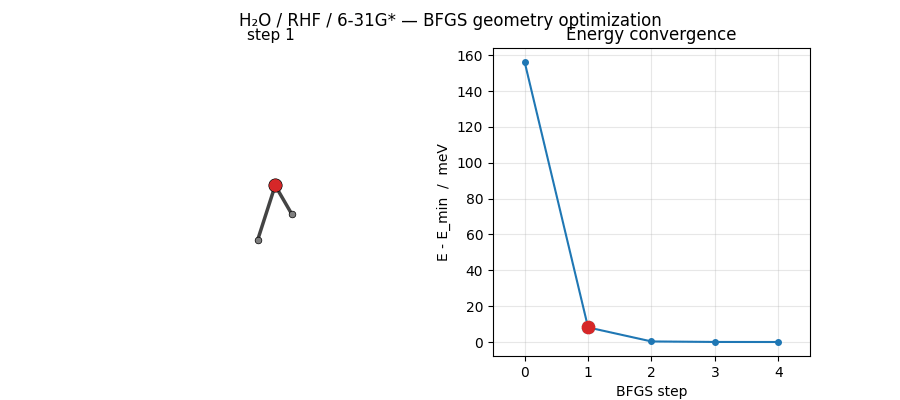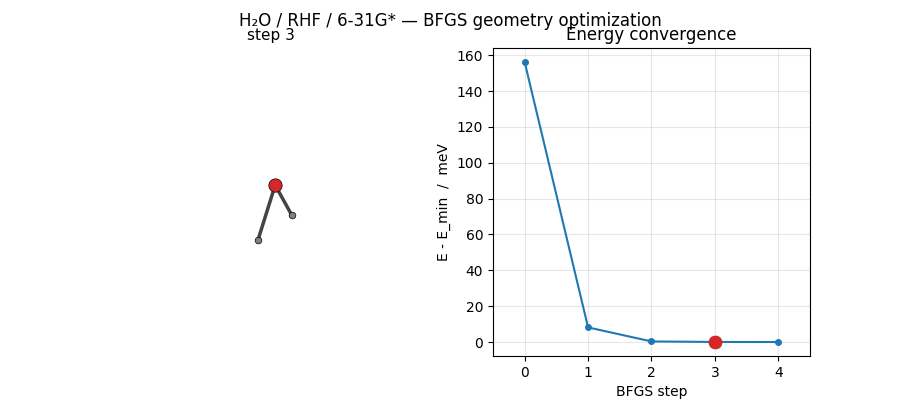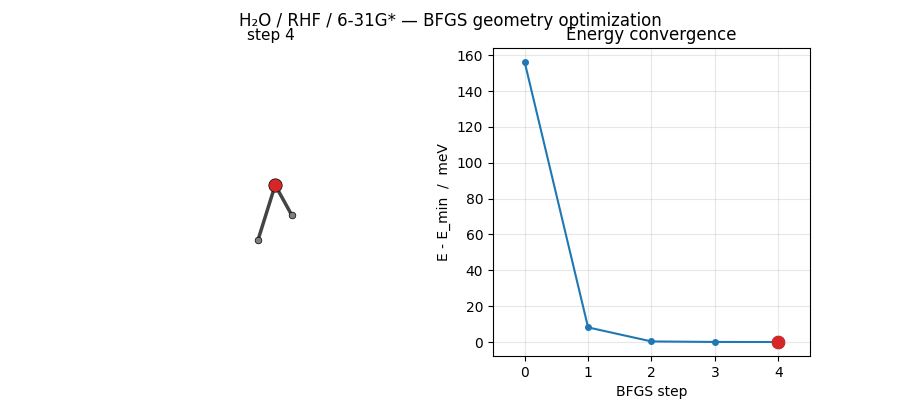
Regenerated by
examples/plots/h2o-opt-trajectory-animation.py
from the per-step .traj file. Pattern is the same as the
normal-mode animation in tutorial 9:
matplotlib FuncAnimation + PillowWriter, the only difference is
that the input here is a series of saved-during-optimization
positions rather than displaced-along-mode positions.
The .traj file holds positions, energies, and forces for every
frame. ASE’s built-in GUI animates it interactively:
ase gui water_opt.traj
Or inspect programmatically:
import numpy as np
from ase.io import read
frames = read("water_opt.traj", index=":")
energies = np.array([f.get_potential_energy() for f in frames])
print(f"Energy dropped {energies[0] - energies[-1]:.4f} eV over "
f"{len(frames) - 1} steps")
DFT geometries¶
Same call, different method:
run_job(
mol, basis="def2-tzvp", method="rks", functional="PBE",
output="water_pbe_opt",
optimize=True,
)
PBE lengthens O-H bonds by ~0.02 Å relative to HF and pulls the HOH angle back toward 104°, generally matching experiment more closely (with the well-known caveat that GGA over-binds H-bonds somewhat).
Theory¶
What “optimization” means on a Born-Oppenheimer PES¶
The Born-Oppenheimer approximation decouples nuclear and electronic motion: fix the nuclei at positions \(\mathbf{R} = \{\mathbf{R}_A\}\), solve the electronic Schrödinger equation for those positions, and treat the resulting electronic energy \(E(\mathbf{R})\) as a potential for the nuclei. Geometry optimization is the problem of finding a stationary point on this potential-energy surface (PES):
A local minimum has all positive eigenvalues of the Hessian \(\mathbf{H}_{AB} = \partial^2 E / \partial \mathbf{R}_A \partial \mathbf{R}_B\); a transition state has exactly one negative eigenvalue.
The Hellmann-Feynman theorem and analytic forces¶
For a variational electronic method at convergence, the forces \(\mathbf{F}_A = -\mathbf{g}_A\) reduce to expectation values of explicit derivatives of the Hamiltonian:
This is the Hellmann-Feynman theorem. For an atom-centered Gaussian basis the orbitals themselves move with the nuclei, so the full analytic gradient also needs Pulay forces — corrections from the basis-function derivatives. vibe-qc assembles all the pieces (one-electron derivatives, two-electron ERI derivatives, DFT-grid derivatives for KS, and dispersion-gradient contributions) into an analytic \(\mathbf{g}_A\) at every SCF step. Accuracy is bounded by the SCF tolerance: default settings give forces better than \(10^{-8}\) Ha/bohr.
BFGS: quasi-Newton in quasi-reality¶
A Newton step along the PES would be \(\Delta \mathbf{R} = -\mathbf{H}^{-1} \mathbf{g}\), but computing the Hessian is expensive (tutorial 09 will do it by finite differences). Quasi-Newton methods build an approximate inverse Hessian \(\mathbf{B}_k\) from the history of steps and gradients. BFGS (Broyden-Fletcher- Goldfarb-Shanno) is the canonical update: given step \(\mathbf{s}_k = \mathbf{R}_{k+1} - \mathbf{R}_k\) and gradient change \(\mathbf{y}_k = \mathbf{g}_{k+1} - \mathbf{g}_k\),
Starting from \(\mathbf{B}_0 = \alpha \mathbf{I}\) (a loose diagonal model), BFGS accumulates curvature information so the quadratic model becomes genuinely predictive near the minimum.
The line-search variant used here¶
vibe-qc’s run_job(..., optimize=True) actually runs ASE’s
BFGSLineSearch, not bare BFGS. The line search enforces the
Wolfe conditions — sufficient-decrease plus curvature — on each
step, so every accepted move reduces the energy. This matters on
flat / weakly-bound surfaces (H-bonded clusters, van-der-Waals
complexes) where plain BFGS’s quadratic model sometimes predicts
wild steps that blow the system apart; the line search catches
them. Cost: 1–3 extra SCFs per BFGS step when the first trial
step is uphill.
Convergence criteria¶
vibe-qc stops when the maximum-component force is below
fmax (units eV/Å, ASE convention). Default fmax = 0.05 is
“medium-tight” (~1 × 10⁻³ Ha/bohr). Benchmark-quality geometries use
fmax = 0.01; H-bonded clusters often need fmax = 0.2 because
force noise from the DFT grid dominates below that floor.
Resources¶
~150 MB peak RAM, ~5 s on one core (Apple M2 baseline) for the
H₂O example: ~10 BFGSLineSearch steps, each one HF/6-31G* +
analytic gradient. The water-trimer optimization in
examples/molecular/input-h2o-trimer-opt.py is ~5 minutes — basis size
and step count both grow with system size.
References¶
Born-Oppenheimer approximation. M. Born and J. R. Oppenheimer, “Zur Quantentheorie der Molekeln,” Ann. Phys. 389, 457 (1927).
Hellmann-Feynman theorem. H. Hellmann, Einführung in die Quantenchemie, Deuticke (1937); R. P. Feynman, “Forces in molecules,” Phys. Rev. 56, 340 (1939).
Pulay forces. P. Pulay, “Ab initio calculation of force constants and equilibrium geometries in polyatomic molecules. I. Theory,” Mol. Phys. 17, 197 (1969).
BFGS update. C. G. Broyden, “The convergence of a class of double-rank minimization algorithms,” IMA J. Appl. Math. 6, 76 (1970) and parallel papers by Fletcher, Goldfarb, Shanno (1970). Textbook treatment: J. Nocedal and S. J. Wright, Numerical Optimization, 2nd ed., Springer (2006), chapters 6–8.
Wolfe conditions / line search. P. Wolfe, “Convergence conditions for ascent methods,” SIAM Review 11, 226 (1969).
ASE (the driver). A. H. Larsen et al., “The atomic simulation environment — a Python library for working with atoms,” J. Phys. Condens. Matter 29, 273002 (2017).
Next¶
Post-SCF analysis — charges, bond orders, and dipole moment at the optimized geometry.
Vibrational frequencies — once the geometry is at a stationary point, compute the Hessian to classify it.