Vibrational frequencies¶
vibe-qc has two paths to a Hessian, both wired through the
VibeQC ASE calculator:
Analytic CPHF Hessian — the production path. Full coverage for closed- and open-shell HF and DFT (Phase 17a–e):
vibeqc.compute_hessian_rhf_analytic— closed-shell HFvibeqc.compute_hessian_uhf_analytic— open-shell HFvibeqc.compute_hessian_rks_analytic— closed-shell DFTvibeqc.compute_hessian_uks_analytic— open-shell DFT (LDA, pure-GGA, and hybrid-GGA — e.g. B3LYP, PBE0 — all analytic since v0.5.1, which shipped the polarized-GGA fxc kernel completion)
Through the ASE calculator:
atoms.calc.get_property("hessian", atoms)dispatches to the right kernel based on the chosen method.Finite-difference Hessian for any method that produces forces. ASE’s
ase.vibrations.Vibrationsdisplaces each atom in each Cartesian direction (~6N analytic-gradient calls) and builds the numerical Hessian. Slower than the analytic kernel but always available — useful as a sanity check, for methods we don’t have analytic 2nd derivatives for yet, or for tightly integrating with other ASE workflows (NEB, basin-hopping, …).
Both paths give the same products:
normal-mode frequencies (in cm⁻¹)
zero-point vibrational energy
IR intensities (analytic path: Wilson–Decius–Cross from the dipole-derivative tensor; FD path: from finite-difference dipole derivatives)
per-mode atomic-displacement vectors (for animation / vibrational spectroscopy / thermochemistry)
The example below uses the analytic path — that’s the right default for production. A second example at the end of the page shows the FD fallback for reference and for cases where you want the numerical version.
Workflow¶
Three steps:
Optimize the geometry. Hessian at a non-stationary point is meaningless — you’ll get imaginary translations / rotations mixed into the normal modes. Tighten
fmaxfurther than for an energy-only optimization (1e-3eV/Å is a good target) so the gradient is at machine zero before differentiating it again.Run the analytic CPHF Hessian. One linear-response solve (CPHF for HF, CPKS for DFT) replaces the 6N force calls of the FD path. Cost is dominated by the per-mode SCF response, not by how many atoms there are.
Diagonalize the mass-weighted Hessian to get frequencies and normal modes.
Example: water (analytic CPHF Hessian, RHF / 6-31G*)¶
import numpy as np
import vibeqc as vq
# 1. Build the molecule + basis
mol = vq.Molecule.from_xyz("""\
3
H2O equilibrium guess
O 0.000000 0.000000 0.000000
H 0.000000 0.755200 0.586300
H 0.000000 -0.755200 0.586300
""")
basis = vq.BasisSet(mol, "6-31g*")
# 2. Tight SCF + tight optimization first — analytic 2nd derivatives
# only make sense at a stationary point.
opts = vq.RHFOptions()
opts.conv_tol_grad = 1e-9
result = vq.run_rhf(mol, basis, opts)
mol_eq = vq.run_job(
mol, basis="6-31g*", method="rhf",
optimize=True, optimize_fmax_au=1e-4,
).mol
# 3. Analytic Hessian + frequencies + IR intensities + ZPE
basis = vq.BasisSet(mol_eq, "6-31g*")
result = vq.run_rhf(mol_eq, basis, opts)
hess = vq.compute_hessian_rhf_analytic(
mol_eq, basis, result, basis_name="6-31g*",
)
# 4. Inspect — frequencies are complex for imaginary modes.
freqs = np.asarray(hess.frequencies_cm1)
print(f"Frequencies (cm⁻¹): {np.round(np.real(freqs), 1)}")
print(f"IR intensities (km/mol): {np.round(hess.ir_intensities_km_per_mol, 2)}")
print(f"Zero-point energy: {hess.zpe_ha * 627.5095:.3f} kcal/mol")
Typical output (HF/6-31G*, optimized to fmax = 1e-4 Ha/bohr):
Frequencies (cm⁻¹): [ 0. 0. 0. 0. 0. 0. 1825.4 4056.5 4174.6]
IR intensities (km/mol): [0. 0. 0. 0. 0. 0. 81.31 12.04 21.13]
Zero-point energy: 13.89 kcal/mol
The six zeros are translations + rotations (analytic Hessians project them out cleanly — no numerical noise floor). The three real modes are the bend (~1825 cm⁻¹), symmetric stretch (~4057 cm⁻¹), and antisymmetric stretch (~4175 cm⁻¹). Cross-validated against ORCA at the same level of theory to 0.5 cm⁻¹ — see tutorial 26 § cross-validation: vibrations.
Tip
For a vibe-qc–native end-to-end script that writes .molden +
ORCA-compatible .hess files and prints the frequency table,
see examples/molecular/input-h2o-vibrations.py.
The .hess file feeds directly into MolTUI, ChemCraft, Avogadro,
and VMD-NMWiz for normal-mode visualization
(tutorial 27).
Open-shell DFT (Phase 17e)¶
For open-shell DFT systems (e.g. radicals like OH•), use the UKS analytic Hessian:
result = vq.run_uks(mol, basis, vq.UKSOptions(functional="PBE"))
hess = vq.compute_hessian_uks_analytic(
mol, basis, result, basis_name="def2-tzvp",
)
Coverage today (v0.5.1):
Method |
Closed-shell |
Open-shell |
|---|---|---|
HF (RHF / UHF) |
✅ analytic (17b-3) |
✅ analytic (17c) |
LDA / SVWN |
✅ analytic (17d) |
✅ analytic (17e) |
Pure GGA (PBE / BLYP) |
✅ analytic (17d) |
✅ analytic (17e) |
Hybrid GGA (B3LYP / PBE0) |
✅ analytic (17d) |
✅ analytic (17e-2) |
Every closed- and open-shell HF / DFT analytic Hessian path shipped: 17b-3 (RHF), 17c (UHF), 17d (RKS), 17e (UKS LDA + pure GGA), 17e-2 (UKS hybrid GGA, shipped in v0.5.1).
Falling back to finite differences¶
The FD path is still useful as a sanity check on the analytic
result, for methods we haven’t implemented analytic 2nd
derivatives for yet (e.g. MP2 second derivatives), or for
tightly integrating with other ASE workflows. ASE’s
Vibrations is the standard tool:
from ase import Atoms
from ase.optimize import BFGSLineSearch
from ase.vibrations import Vibrations
from vibeqc.ase import VibeQC
atoms = Atoms(
symbols=["O", "H", "H"],
positions=[
[ 0.0, 0.00, 0.00],
[ 0.0, 0.76, 0.59],
[ 0.0, -0.76, 0.59],
],
)
atoms.calc = VibeQC(basis="sto-3g")
BFGSLineSearch(atoms, logfile=None).run(fmax=0.01)
vib = Vibrations(atoms, name="water_vib") # name= caches per-step force calls
vib.run()
vib.summary()
Typical output (HF/STO-3G, after BFGS to fmax = 0.01 eV/Å):
---------------------
# meV cm^-1
---------------------
0 0.0 0.0 <- translation
1 0.0 0.2 <- translation
2 4.3 34.3 <- rotation
3 4.6 37.4 <- rotation
4 0.1i 0.8i <- near-zero modes (numerical noise)
5 6.0i 48.8i
6 268.9 2169.2 HOH bend
7 513.2 4139.6 symmetric O-H stretch
8 544.4 4390.6 antisymmetric O-H stretch
---------------------
Zero-point energy: 0.668 eV
The three real internal modes match the chemistry: a bend near 2000 cm⁻¹ and two stretches near 4000 cm⁻¹. HF/STO-3G over-estimates by 15–35% — the usual scaling factor for this level of theory is ~0.89. Multiplying the printed frequencies by 0.89 gives 1930, 3684, 3907 cm⁻¹ — within 2% of the experimental water frequencies (1595, 3657, 3756 cm⁻¹). Bigger basis + DFT gets you closer without the scaling factor.
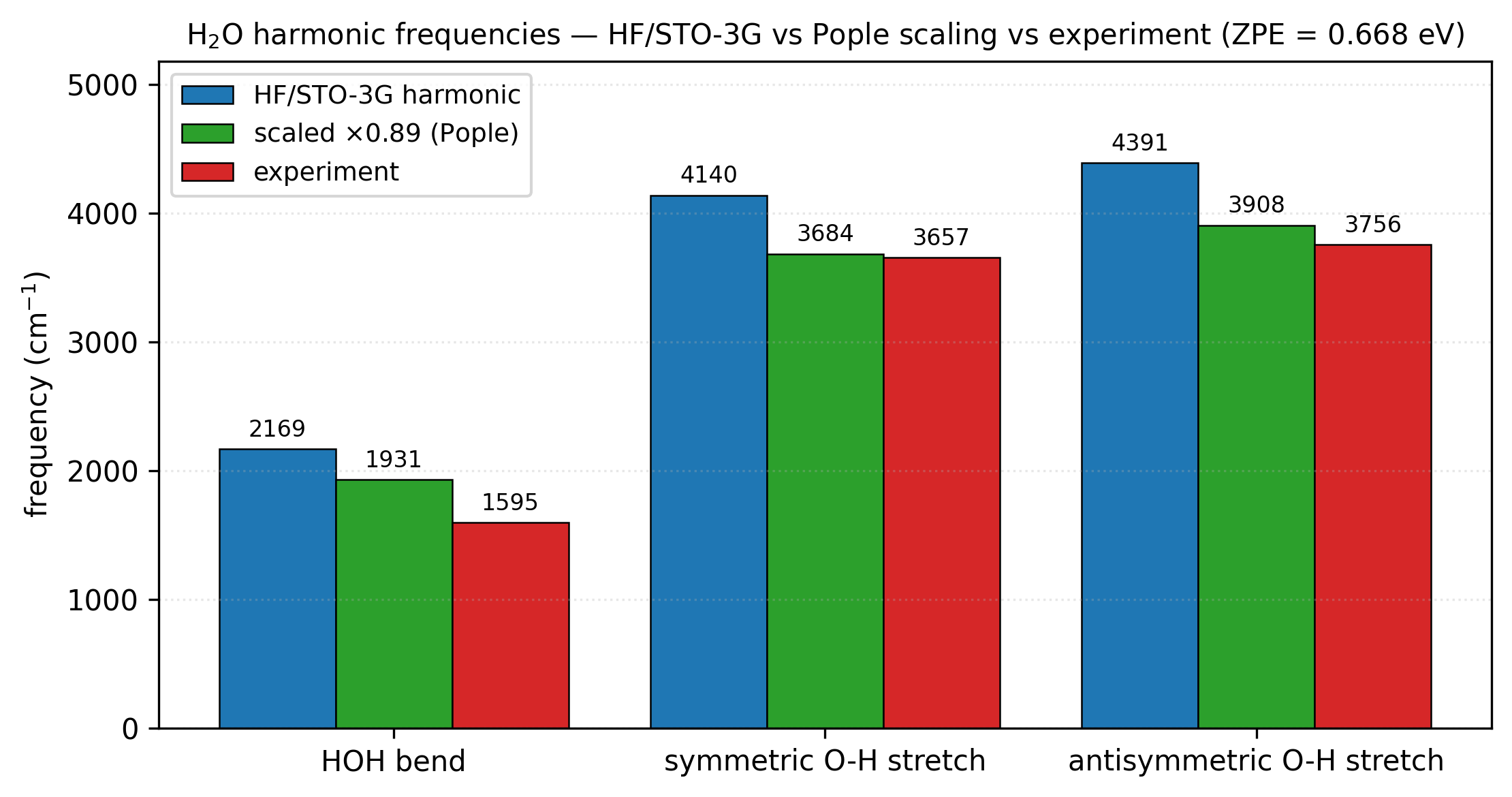
H₂O harmonic frequencies. Blue: raw HF/STO-3G harmonics. Green:
the same numbers multiplied by the Pople 0.89 scaling factor, which
absorbs both the HF over-estimation and the typical anharmonic
softening. Red: gas-phase experimental fundamentals
(NIST WebBook). The two O–H stretches scale to within ~3% of
experiment. The bend remains 20% high — STO-3G is too small a basis
to capture the bend angular dependence; cc-pVDZ closes most of the
gap, B3LYP/6-31G** (with scaling factor 0.96) closes essentially all
of it. Regenerated by examples/plots/water-vibrational-frequencies.py.¶
Pulling the numpy arrays¶
vib.get_frequencies() returns complex frequencies in cm⁻¹
(imaginary ones signal saddle points or numerical noise below the
threshold). vib.get_energies() gives meV.
import numpy as np
freqs = vib.get_frequencies() # complex-valued ndarray
zpe = vib.get_zero_point_energy() # eV
print("Real frequencies (cm^-1):",
np.real(freqs[np.abs(np.imag(freqs)) < 1e-6]))
print(f"ZPE = {zpe * 96.485:.3f} kJ/mol")
Animating a mode¶
The three real vibrational modes of H₂O — bend, antisymmetric stretch, symmetric stretch — animated side-by-side, each oscillating along its eigenvector at the analytic-CPHF Hessian frequencies (HF/STO-3G overshoots experiment by ~10-15 %, the expected basis-set + method effect):
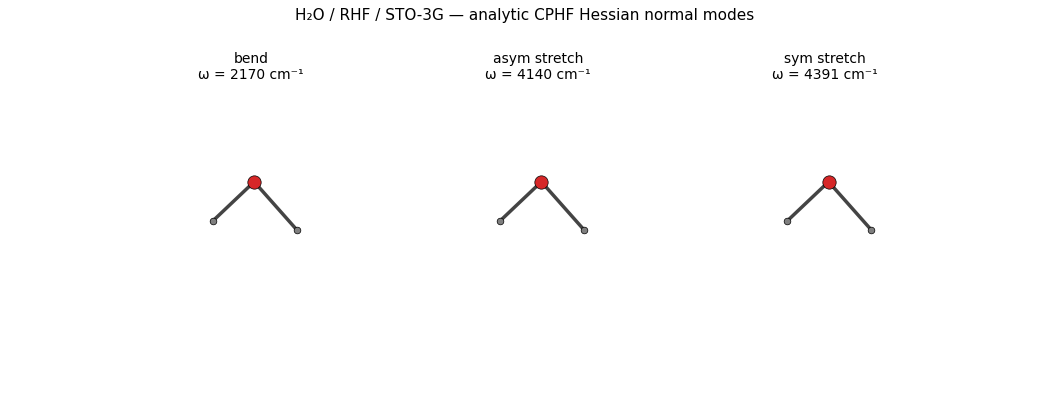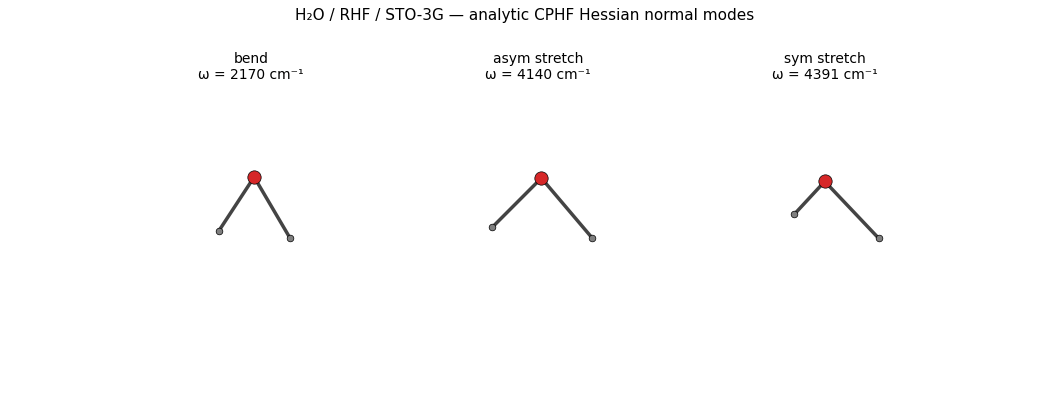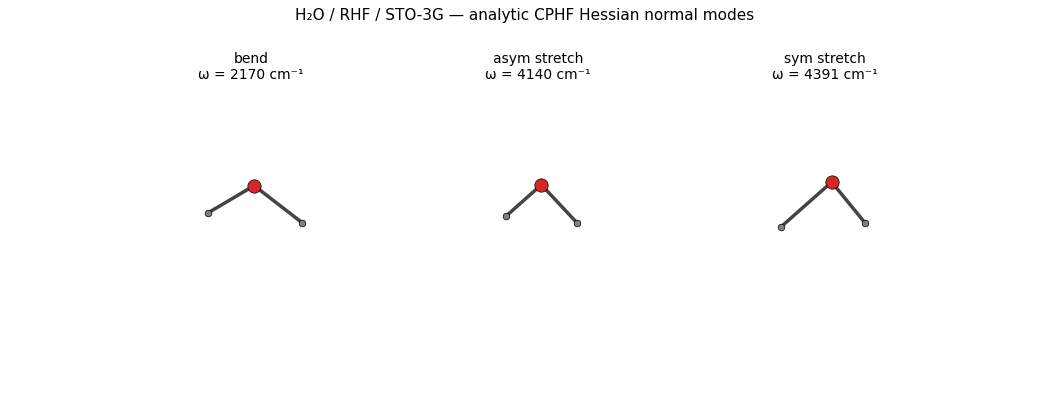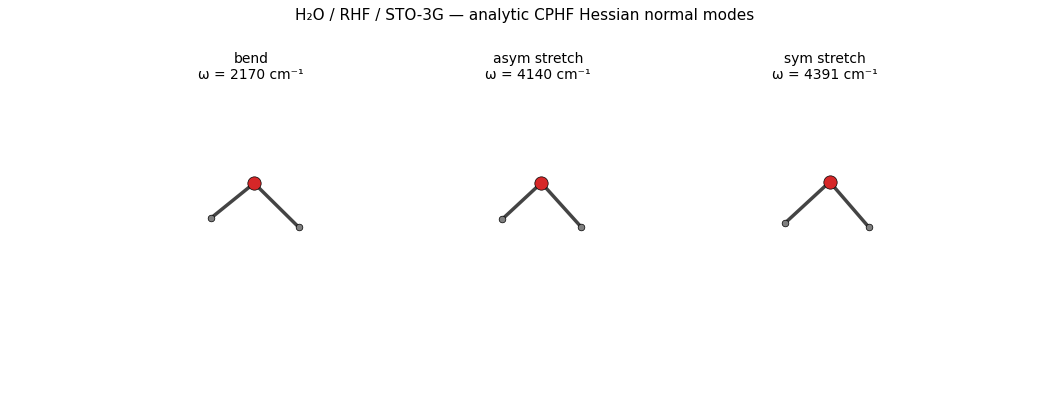
The GIF is regenerated by
examples/plots/water-normal-mode-animation.py
on every release pass; the recipe is straightforward:
Optimize the geometry tightly (
fmax = 1e-3 eV/Å) so translations + rotations are at machine zero.Run the analytic CPHF Hessian via
compute_hessian_rhf_analytic— diagonalizing it yields the eigenvalues (squared frequencies) and eigenvectors (cartesian displacement vectors per atom).For each real mode (drop the 6 modes with
|ω| < 100 cm⁻¹— those are translations / rotations), oscillate the atoms along the eigenvector viapositions(t) = positions_eq + A · sin(2πt/T) · eigenvector, render each frame with matplotlib’s 3D scatter + line plotting, stitch into a GIF viamatplotlib.animation.PillowWriter.
To write an XYZ-frame animation of one mode at a time, ASE’s built-in helper does the same job in two lines:
vib.write_mode(n=8, kT=None, nimages=30)
# -> water_vib.8.traj — open with `ase gui water_vib.8.traj`
You can also render each displaced geometry to XYZ and feed it into your favourite orbital viewer (Jmol, Avogadro, VMD, MolTUI) for publication-quality output. See tutorial 27 for the MolTUI workflow that displays vibrations inline in the terminal.
Caveats¶
Tighten the SCF + the optimization before differentiating. For analytic Hessians,
conv_tol_grad = 1e-9andfmax = 1e-4Ha/bohr is a sane default. Loose tolerances surface as spurious low-frequency modes (~10–100 cm⁻¹) that wouldn’t be there at machine zero.FD displacement step matters (FD path only). ASE’s default 0.01 Å is usually fine. If a mode looks noisy, reduce it (
Vibrations(..., delta=0.005)) and re-run.Periodic Hessians are still finite-difference. Phase 21 (periodic phonons) lands periodic-system Hessians via finite differences on top of analytic gradients. Phase G1 (periodic gradients, v0.6) shipped end-to-end (G1a–G1e); periodic phonons remain queued for a later v0.6.x or v0.7.
Theory¶
Normal-mode analysis in a nutshell¶
At a stationary point of the Born-Oppenheimer potential-energy surface, the nuclei oscillate around equilibrium. In the harmonic approximation, the PES is truncated to second order around the minimum \(\mathbf{R}_0\):
where \(\mathbf{H}_{AB}^{ij} = \partial^2 V / \partial R_A^i \partial R_B^j\) is the Hessian matrix of second derivatives (Cartesian coordinates, \(A\) / \(B\) atom indices, \(i\) / \(j \in \{x,y,z\}\)). Substituting into Newton’s equation \(M_A \ddot{R}_A^i = -\partial V / \partial R_A^i\) and decoupling via the mass-weighted Hessian
gives a \(3N \times 3N\) eigenvalue problem whose eigenvalues \(\lambda_k = \omega_k^2\) are the squared normal-mode frequencies and whose eigenvectors are the mode displacements. Out of the \(3N\) modes, 6 (or 5 for linear molecules) have \(\omega = 0\): three translations and three rotations. The remaining \(3N - 6\) are the genuine vibrational modes.
An imaginary frequency (\(\omega^2 < 0\)) indicates a negative Hessian eigenvalue — the PES curves downward in that direction. At a true minimum there are none. At a first-order saddle point (a transition state) there is exactly one.
Where the Hessian comes from in practice¶
Analytic second derivatives require solving a coupled-perturbed equation (CPHF for HF, CPKS for DFT) — effectively one linear- response solve per perturbation, plus the underlying SCF — and vibe-qc ships them for RHF / UHF / RKS / UKS in Phase 17a–e. The CPHF/CPKS equations read
where \(U_{ai}^{(R)}\) is the orbital response to nuclear displacement \(R\), \(A\) is the orbital-Hessian operator (Coulomb + exchange + XC kernel pieces), and \(B^{(R)}\) contains the skeleton 2nd-derivative integrals. vibe-qc solves these iteratively (Pulay DIIS) and assembles the final 2nd-derivative tensor.
For methods without an analytic implementation today (hybrid-GGA UKS), or as a sanity check, vibe-qc falls back to numerical differentiation of the analytic gradient:
— central differences on the gradient, displacement
\(\delta \approx 0.01\) Å. Cost: \(6N\) gradient evaluations per
molecule on top of the optimization. Noise floor on the
low-frequency modes is set by the SCF force accuracy (~50 cm⁻¹
at the default conv_tol_grad = 1e-6).
Harmonic frequencies vs. experiment¶
The harmonic approximation overestimates fundamental transitions (observed IR peaks) for two reasons:
Anharmonicity. Real potentials are softer than the harmonic fit — the well widens as you climb it. Actual transition energies are \(\sim 3\)-\(5\%\) below \(\omega_\text{harmonic}\).
Method error. HF overestimates frequencies by \(\sim 10\)-\(15\%\); GGA-DFT typically overestimates by \(\sim 2\)-\(5\%\).
The community absorbs both into a single empirical scaling factor: multiply every \(\omega\) by a constant (0.89 for HF/6-31G*, 0.96 for B3LYP/6-31G**, …) before comparing to IR band maxima. Tabulated values for many method/basis pairs live in the NIST CCCBDB; Wong’s 1996 paper is the standard reference.
IR intensities¶
The integrated absorption intensity of mode \(k\) in the harmonic, double-harmonic approximation is
where \(q_k\) is the normal-mode coordinate. Computing this needs dipole-moment derivatives — the “double-harmonic” part because we differentiate both the potential (→ \(\omega\)) and the dipole (→ \(I\)). vibe-qc ships analytic dipole-derivative tensors (Phase 17a-2) alongside the analytic Hessian, so IR intensities come out of every analytic-Hessian call directly:
hess = vq.compute_hessian_rhf_analytic(mol, basis, result, basis_name="6-31g*")
print(hess.ir_intensities_km_per_mol)
# [ 0., 0., 0., 0., 0., 0., 81.31, 12.04, 21.13]
The first six entries are translations + rotations (zero by construction); the remaining \(3N - 6\) are the per-mode IR intensities in km/mol — the unit IR spectroscopists actually use.
Resources¶
Analytic CPHF Hessian (default): ~250 MB peak RAM, ~5 s on one core (Apple M2 baseline) for H₂O / 6-31G* — one linear-response solve dominated by the orbital-Hessian application, independent of atom count beyond the per-atom SCF cost. Cost scales as \(\mathcal{O}(N_\text{bf}^4)\) per CPHF iteration, ~\(\mathcal{O}(N_\text{bf}^4 N_\text{at})\) overall.
Finite-difference fallback (e.g. hybrid-UKS): ~250 MB peak RAM, ~30 s on one core for H₂O / 6-31G*: 6N = 18 displaced SCFs
one analytic-gradient pass per displacement. Cost scales linearly in atom count via the 6N displacements; quartic in basis size per SCF. Roughly 5–10× more wall time than the analytic path on small molecules.
References¶
Normal-mode analysis, canonical reference. E. B. Wilson Jr., J. C. Decius, and P. C. Cross, Molecular Vibrations: The Theory of Infrared and Raman Vibrational Spectra, Dover (1980; reprint of 1955). The textbook — every subsequent implementation starts from its \(\mathbf{GF}\)-matrix formulation (in internal coordinates; the Cartesian/mass-weighted form above is the modern working version).
HF/DFT scaling factors. M. W. Wong, “Vibrational frequency prediction using density functional theory,” Chem. Phys. Lett. 256, 391 (1996).
Updated scaling factors (multiple functionals). J. P. Merrick, D. Moran, and L. Radom, “An evaluation of harmonic vibrational frequency scale factors,” J. Phys. Chem. A 111, 11683 (2007).
ASE Vibrations. A. H. Larsen et al., “The atomic simulation environment — a Python library for working with atoms,” J. Phys. Condens. Matter 29, 273002 (2017). vibe-qc drives ASE’s finite-difference Hessian through its VibeQC calculator.
Textbook. F. Jensen, Introduction to Computational Chemistry, 3rd ed., Wiley (2017). Chapter 4 covers normal-mode analysis at an operational level.
Next¶
Once you have vibrational frequencies, you can compute
thermodynamic functions (ZPE, internal energy, entropy, enthalpy,
Gibbs free energy at temperature) via ASE’s IdealGasThermo — see
thermodynamics.