The QVF file format, end to end¶
You will learn: what a .qvf archive is, the two ways vibe-qc
produces one, the anatomy of its manifest, and how to write and read
the main section-kind families, structure, scalar fields, the full
wavefunction, spectra, electronic bands and density of states, optimisation
and reaction paths, vibrations, and the provenance / citation metadata. By the
end you will have produced three representative archives and validated them.
The full current writer / viewer kind matrix lives in the
QVF design doc; newer
kinds such as basis.ao, scan.surface, COOP/COHP, Fermi surfaces, phonons,
EOS, and QTAIM have dedicated examples or producer paths outside this compact
showcase.
This is the format companion to
vibe-view: an end-to-end walkthrough, which walks the
vibe-view GUI. The vibe_view_walkthrough tutorial shows what a
routine job emits; this tutorial shows the whole format surface and
the lower-level writer API you reach for when you want to put something
in a .qvf that run_job does not emit on its own.
Prerequisites:
vibeqcinstalled and a working SCF setup (any Fundamentals tutorial).Optional:
vibe-viewinstalled (for the read-back and GUI steps),pip install -e 'vibe-view/[viewer]'from the checkout root.
Time to complete: about 15 minutes.
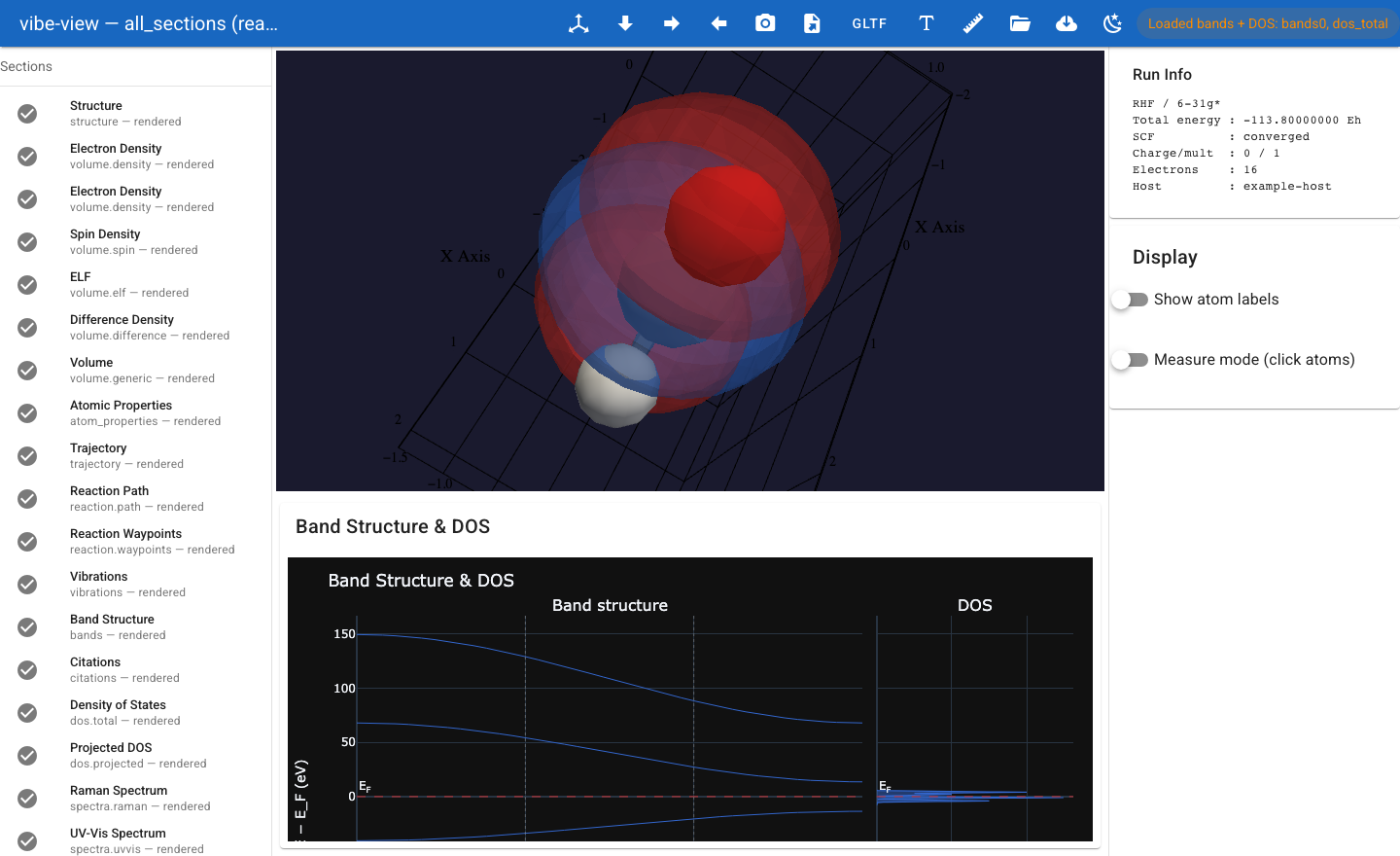
One QVF archive opened in vibe-view — a bands + dos.total figure on a
shared energy axis. By the end of this tutorial you will have written and
validated a representative multi-section .qvf bundle.¶
1. What a .qvf is¶
A .qvf (Quantum Visualization Format) is a single ZIP
archive that bundles everything a calculation produced for
visualization, geometry, scalar fields, spectra, bands, trajectories,
provenance, instead of scattering it across .cube, .xyz,
.molden, .xsf and log files. One file you can hand to a colleague,
attach to a paper’s SI, or open in vibe-view.
Inside, it is just a ZIP with three layers:
water.qvf
├── manifest.json # the index: every section + member, with checksums
├── structure/structure.json # JSON members (small, human-readable)
├── volumes/density.dat # binary members (large numeric arrays)
├── volumes/density_grid.json
├── ...
manifest.jsonis the single source of truth. It lists every section (a unit of content like “the electron density” or “the IR spectrum”), and within each section the members (the files that carry the bytes), each with apath, aformat(jsonorbinary), and asha256checksum.JSON members carry small structured data (atom lists, spectrum peaks, grid descriptors).
Binary members carry large numeric arrays (volumetric grids, MO coefficients, trajectory coordinates) as raw little-endian buffers, with their
dtypeandshapedeclared in the manifest.
The canonical contract is the JSON Schema at
python/vibeqc/output/formats/qvf_manifest.schema.json; the full
specification (units, section kinds, the validation and extension
models) is the QVF design doc.
The write-time validation gate
write_qvf validates the archive it just wrote against the canonical
schema before returning, and if validation fails it deletes the
file and raises. A .qvf on disk that came from vibe-qc is therefore
guaranteed schema-valid. This is also why the showcase script in this
tutorial is a useful bug-finder: writing one archive per section kind
and getting no exception is the test that every writer path is sound.
2. Two ways to produce a .qvf¶
There are two routes to an archive: the high-level output_qvf=True flag
on a normal job, which covers almost every case, and the lower-level
write_qvf(...) call for sections a job does not emit on its own.
a. The natural way, output_qvf=True¶
Any run_job / run_periodic_job call emits a .qvf when you pass
output_qvf=True. The sections that appear are whatever the job
computed: ask for cubes and you get volume.*; ask for a Hessian and
you get vibrations + spectra.ir; optimise and you get a
trajectory. This is what vibe-view: an end-to-end walkthrough
uses, and it is what you want 95% of the time:
from vibeqc import Atom, Molecule, run_job
mol = Molecule([
Atom(8, [0.0, 0.00, 0.00]),
Atom(1, [0.0, 1.43, -0.98]),
Atom(1, [0.0, -1.43, -0.98]),
])
run_job(
mol, basis="6-31g*", method="rhf",
optimize=True, # -> trajectory
hessian=True, # -> vibrations + spectra.ir
write_cube=["density", "homo", "lumo"], # -> volume.density + volume.orbital
write_molden_file=True, # -> wavefunction.gto
write_population_file=True, # -> atom_properties
citations=True, # -> citations
output="water", output_qvf=True,
)
b. The explicit way, write_qvf(...)¶
To put something in a .qvf that run_job does not emit, a
difference density you computed yourself, a UV/Vis spectrum from an
external tool, hand-built reaction-path waypoints, call the writer
directly. It takes an OutputPlan and a bag of
context keyword arguments, one per section kind:
from vibeqc.output.formats.qvf import write_qvf, validate_qvf
from vibeqc.output.plan import OutputPlan
plan = OutputPlan.from_run_job_kwargs(
output="custom", method="rhf", basis="sto-3g", functional=None
)
write_qvf(
"custom", plan,
molecule=mol, # -> structure
volume_data={"Density": (rho, origin, span)}, # -> volume.density
raman_data={"frequencies": [...], "intensities": [...]}, # -> spectra.raman
# ... one kwarg per section you want ...
)
The rest of this tutorial walks the representative section families and the
context keys that produce them. The full runnable compact showcase is
examples/vibe_view/showcase_qvf_all_sections.py, it builds three
archives covering the historical 27-kind gate and validates each. Run it now
and refer back to its output as you read:
~/path/to/vibeqc/.venv/bin/python examples/vibe_view/showcase_qvf_all_sections.py
3. Anatomy of the manifest¶
Open any archive and read its manifest, this is the consumer’s entry point:
import zipfile, json
manifest = json.loads(zipfile.ZipFile("water.qvf").read("manifest.json"))
print("QVF v", manifest["qvf_version"])
print("from ", manifest["source"]) # program, version, calculation
print("provenance", manifest.get("provenance")) # method, basis, energy, ...
for s in manifest["sections"]:
print(f" {s['id']:<16s} {s['kind']}")
The manifest root carries:
qvf_version, always1, including for periodic reaction paths, whose per-frame lattices ride as an optional member (see § 8).source, producer program, version, and a calculation string.provenance, best-effort method / functional / basis / charge / multiplicity / SCF energy / convergence (assembled from the context you pass).viewer_defaults, optional producer hints (which section to auto-open, default isovalue / colormap, camera bookmarks).sections, the list. Each entry has anid, akind, optionallabel, and amembersmap of role → member spec. A member spec is{path, format, sha256}for JSON and additionally{dtype, shape}for binary.
All units follow the format’s conventions (positions in Å, grids in bohr, energies in Hartree or eV per the units table).
4. The section kinds, group by group¶
Every snippet below is a context kwarg to write_qvf. The showcase
script assembles them into coherent archives; here they are isolated so
you can see exactly what each kind needs.
4.1 Structure and connectivity¶
The structure kind carries the geometry, with optional bonds and
structure.symmetry members alongside it:
write_qvf(
stem, plan,
molecule=mol, # structure (or system=PeriodicSystem)
bonds_data=[(0, 1, 1.0), (0, 2, 1.0)], # bonds — (i, j, order) triples
symmetry_data={ # structure.symmetry (spglib-style)
"space_group_number": 225,
"space_group_symbol": "Fm-3m",
"point_group": "m-3m",
},
)
structure is the one section almost every archive has. Pass
molecule= for a molecule or system= for a PeriodicSystem (which
adds lattice vectors and pbc flags). bonds is an explicit
connectivity table (omit it and the viewer infers bonds from covalent
radii); structure.symmetry carries the spglib summary.
4.2 Scalar fields, the six volume.* kinds¶
All volumes share the same shape: a dict {label: (data_3d, origin, span)} where data_3d is a 3-D numpy array, origin is the grid
anchor in bohr, and span is the 3×3 matrix of per-voxel step vectors
in bohr.
write_qvf(
stem, plan, molecule=mol,
volume_data={"Electron density": (rho, origin, span)}, # volume.density
mo_data=[...], # volume.orbital
spin_data={"Spin density": (s, origin, span)}, # volume.spin
elf_data={"ELF": (elf, origin, span)}, # volume.elf
generic_volume_data={"RDG": (rdg, origin, span)}, # volume.generic
diff_data={"Δρ": { # volume.difference
"data": diff, "origin": origin, "span": span,
"operand_a": "vol_dens_0", "operand_b": "vol_dens_0",
"description": "ρ(product) − ρ(reactant)",
}},
)
volume.density/volume.spin/volume.elfdiffer only in meaning and the viewer’s default colormap (signed for spin and difference, sequential for density and ELF).volume.genericis the escape hatch for any scalar field that is not one of the named kinds (reduced density gradient, electrostatic potential you computed yourself, …).volume.differencecan link to the twovolume.*sections it was built from viaoperand_a/operand_b(section ids), so a viewer can offer “show me the operands”.mo_data(thevolume.orbitalkind) is a list of per-orbital dicts; theqvf_mo_data(result, basis, molecule, indices)helper builds it for you from a finished SCF (pass the orbital indices to sample), andrun_job(write_cube=[...])produces it automatically.
For the full molecular orbital set without pre-sampling every orbital, use the wavefunction section instead (next).
4.3 The wavefunction, wavefunction.gto¶
The wavefunction.gto kind stores the full GTO basis and MO coefficients
so a viewer can resample any orbital on demand; the qvf_wf_data helper
builds it from a finished SCF:
from vibeqc.output.formats.qvf import qvf_wf_data
write_qvf(stem, plan, molecule=mol,
wf_data=qvf_wf_data(result, basis, mol)) # wavefunction.gto
This embeds the GTO basis (shells, exponents, contraction
coefficients) plus the MO coefficient matrix, so a viewer can resample
any orbital on demand, far cheaper than writing each orbital as its
own volume.orbital. run_job(write_molden_file=True) emits it
automatically. Unrestricted runs carry separate α/β coefficient
members.
Shell coefficients in the archive apply to normalized primitive
Gaussians (see docs/design_qvf_format.md § 4.6 for the formula). The
writer handles this automatically, you pass raw libint shells and
qvf_wf_data does the conversion.
4.4 The spectra family, seven kinds¶
Each spectrum is a small JSON dict of peaks. They share a common
{frequencies, intensities} core; the energy-axis kinds accept
energies_ev and the writer maps it for you.
write_qvf(
stem, plan, molecule=mol,
# spectra.ir is emitted automatically from a Hessian (hessian_result=)
raman_data={"frequencies": fr, "intensities": ri}, # spectra.raman
uvvis_data={"energies_ev": e, "intensities": f}, # spectra.uvvis
ecd_data={"energies_ev": e, "intensities": R}, # spectra.ecd
vcd_data={"frequencies_cm1": fr, "intensities": dA}, # spectra.vcd
nmr_data={"isotope": "1H", "chemical_shifts": [...]}, # spectra.nmr
generic_spectrum_data={ # spectra.generic
"section_id": "xps", "label": "XPS",
"frequencies": [...], "intensities": [...],
},
)
spectra.ir is special: it is derived from a HessianResult (pass
hessian_result=, or use run_job(hessian=True)), not a hand-built
dict, the writer computes the intensities. The other six accept
producer-supplied numbers, which is what you want when the spectrum
came from a method vibe-qc does not yet compute. spectra.generic lets
you name your own axis for anything that does not fit (XPS, a fitted
envelope, …).
Warning
The synthetic spectra in the showcase script are illustrative format-demo data, not computed observables. The format supports carrying these spectra; vibe-qc does not yet compute most of them. Do not cite numbers read out of a demo archive.
4.5 Electronic structure of solids, bands, dos.total, dos.projected¶
The periodic-only kinds carry a band structure along a k-path plus total
and projected densities of states; they need system= rather than
molecule=:
write_qvf(
stem, plan, system=periodic_system,
band_structure=vq.band_structure_hcore(sys, basis, kpath), # bands
dos_data={ # dos.total
"energies": E, "dos": g, "n_spin": 1,
"smearing": 0.1, "fermi_energy_ev": 0.0, "n_electrons": 20.0,
},
pdos_data={ # dos.projected
"energies": E, "projections": g_channels, "n_spin": 1,
"channels": [{"atom_index": 1, "symbol": "O", "l": 1, "label": "O 2p"}],
},
)
bands takes a real BandStructure (the cheapest is
band_structure_hcore, which needs no SCF, see
Band structure and density of states). DOS arrays are [n_points]
(or [2, n_points] for spin-polarized total DOS, [n_channels, n_points] for projected); energies are in eV with the Fermi level at
0. These are the periodic-only kinds, they need system= rather than
molecule=.
Validated periodic chi-CCM-B QVF fixtures¶
The documentation tree also carries two small periodic QVF fixtures from
jk_method="aiccm2026dev-b" that exercise the finite-BvK torus path in
vibe-view. They are useful when checking a consumer’s periodic box, grid,
replication, and vendor-overlay behavior because they contain only precomputed
periodic volume.density / volume.orbital grids, not a periodic
wavefunction.gto section.
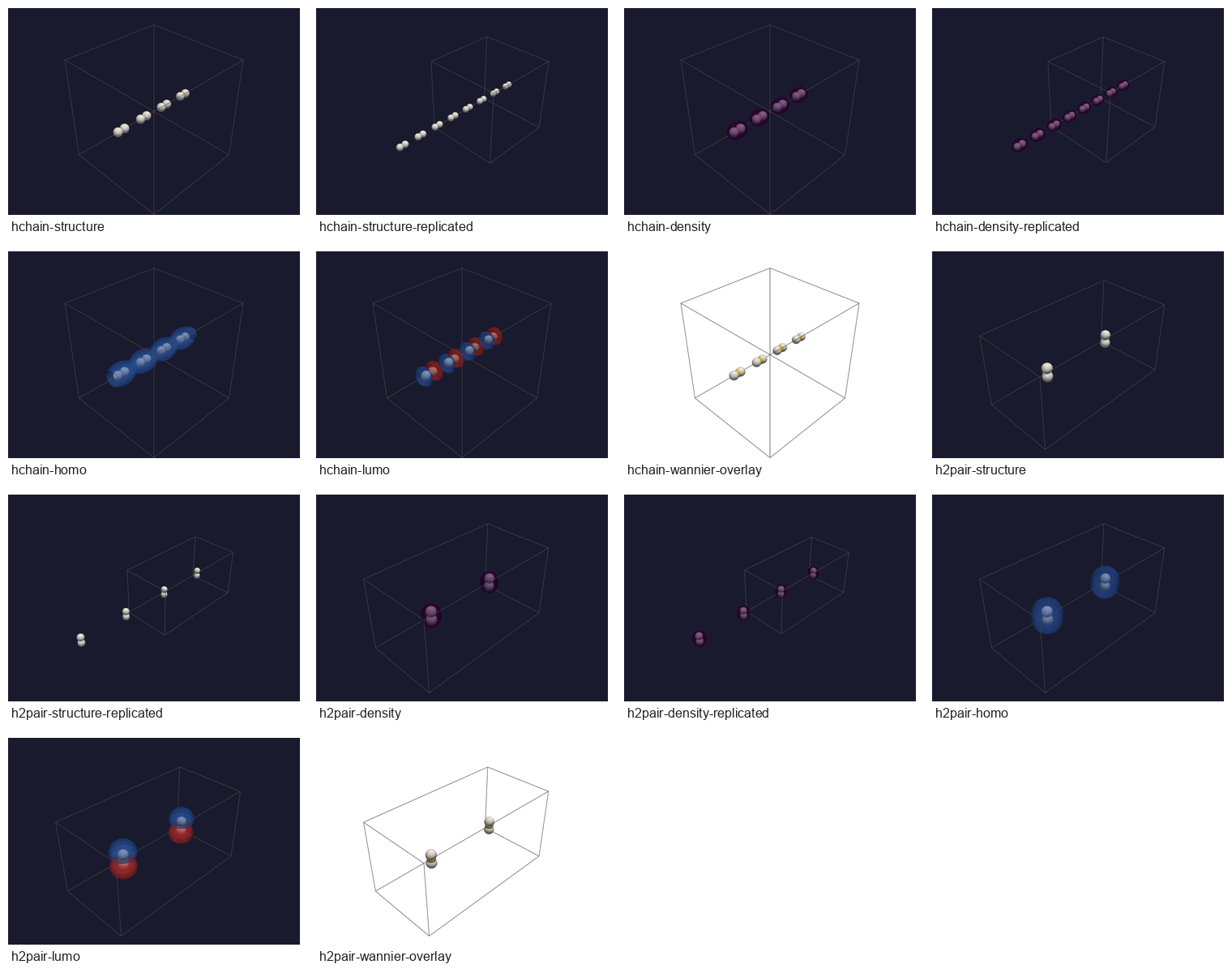
Headless vibe-view captures for the two chi-CCM-B periodic QVF fixtures:
single-cell and replicated structures, density surfaces, orbital surfaces,
and x_ccm.wannier_centers overlays.¶
Case |
Input |
Full output |
System manifest |
QVF archive |
Captures |
|---|---|---|---|---|---|
3D vacuum-padded H-chain, RI, |
structure, replicated structure, density, replicated density, HOMO, LUMO, Wannier overlay |
||||
3D H2-pair, RI, |
structure, replicated structure, density, replicated density, HOMO, LUMO, Wannier overlay |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The vacuum-padded H-chain archive has structure.pbc=[true,true,true],
structure.dimensionality=3, a 40 x 40 x 40 density grid spanning the
full 20 x 20 x 20 bohr BvK supercell, and integrates to 8.001083
electrons. The H2-pair archive has structure.pbc=[true,true,true],
structure.dimensionality=3, a 48 x 24 x 24 grid spanning
24 x 12 x 12 bohr, and integrates to 4.000538 electrons. Both archives
validated with validate_qvf(...) after the public copies were sanitized.
4.6 Paths and dynamics, trajectory, reaction.path, reaction.waypoints, vibrations¶
These kinds carry a stack of geometries (an optimisation or MD run), a typed reaction path or its lightweight waypoint annotation, and normal-mode displacements from a Hessian:
write_qvf(
stem, plan, molecule=mol,
trajectory_frames=frames, # trajectory (list of Molecule)
trajectory_energies=[...],
reaction_waypoints={ # reaction.waypoints (annotates traj0)
"trajectory_ref": "traj0",
"waypoints": [{"frame_index": 0, "label": "start", "kind": "point"}],
},
reaction_path={ # reaction.path (self-contained)
"frames": rxn_frames,
"energies": [...],
"waypoints": [
{"frame_index": 0, "label": "Reactant", "kind": "reactant"},
{"frame_index": 2, "label": "TS", "kind": "transition_state"},
{"frame_index": 4, "label": "Product", "kind": "product"},
],
},
hessian_result=hess, # vibrations (+ spectra.ir)
)
trajectoryis a stack of geometries (a geometry optimisation, an MD run), frames areMoleculeobjects, optionally with per-frame energies.run_job(optimize=True)emits one.reaction.pathis a self-contained path with typed waypoints (reactant/transition_state/intermediate/product/point), exactly what an NEB run produces.reaction.waypointsis the lightweight alternative: instead of re-storing coordinates, it annotates atrajectoryalready in the archive (thetrajectory_refmust name atrajectorysection in the same file, or the writer raises).vibrationscarries normal-mode displacements from aHessianResult; passing one also emitsspectra.ir.
4.7 Provenance and metadata, atom_properties, scf_history, citations, viewer_defaults¶
The metadata kinds carry per-atom charges, the per-iteration SCF history, the embedded BibTeX bundle, and the manifest-root viewer hints:
write_qvf(
stem, plan, molecule=mol,
population_summary=pop, # atom_properties (Mulliken/Löwdin)
scf_history_data=[ # scf_history
{"iter": 0, "energy_eh": -74.9, "delta_e": 1.0, "diis_error": 0.5},
],
bibtex_content=open("water.bibtex").read(), # citations
viewer_defaults={ # manifest-root hints
"auto_open": ["vol_dens_0"],
"vol_dens_0": {"isovalue": 0.05, "colormap": "viridis"},
},
)
atom_properties carries per-atom Mulliken / Löwdin charges;
scf_history the per-iteration energy and DIIS error;
citations the embedded BibTeX bundle
(the same bytes as the .bibtex sidecar, see
Auto-citations: from .out to manuscript bibliography). viewer_defaults is not a
section but a manifest-root block of hints the viewer honours on load.
5. Run the showcase, three representative archives¶
The script produces:
Archive |
Built by |
Kinds it contributes |
|---|---|---|
|
real |
structure, volume.density, volume.orbital, wavefunction.gto, trajectory, vibrations, spectra.ir, atom_properties, scf_history, citations |
|
explicit |
volume.spin / elf / difference / generic, spectra.raman / uvvis / ecd / vcd / nmr / generic, structure.symmetry, bonds, reaction.path, reaction.waypoints, viewer_defaults |
|
explicit |
bands, dos.total, dos.projected, periodic structure, structure.symmetry, periodic reaction.path |
The final lines of its output are the coverage gate:
Coverage gate:
kinds covered: 27/29
...
All 27 implemented section kinds written, validated, and round-tripped (basis.ao + scan.surface exercised separately).
The numbers above are the historical compact-showcase gate for those three
archives, not the full current QVF registry. If one of those writer paths
regresses, the script dies at the offending write_qvf call (the validation
gate) or at the coverage assertion, which is exactly the bug-finding behaviour
we want from it.
6. Validate and read back¶
You do not need vibe-view to consume a .qvf, it is a documented ZIP.
The producer-side gate already validated it, but you can re-check:
from vibeqc.output.formats.qvf import validate_qvf
report = validate_qvf("crystal.qvf")
assert report["valid"], report["errors"]
To read it with nothing but the standard library, verifying checksums
yourself, then pulling a binary array out, follow the
QVF consumer reference. The essential
move for a binary member is np.frombuffer(zf.read(m["path"]), dtype=m["dtype"]).reshape(m["shape"]). The showcase script’s
_verify_one_checksum does the SHA-256 round-trip in a few lines.
If vibe-view is installed, its reader gives you the same manifest plus classification of which kinds it can render:
from vibeview.qvf import QVFReader
reader = QVFReader("spectroscopy.qvf")
print(len(reader.manifest["sections"]), "sections")
7. Open it in vibe-view¶
If vibe-view is installed, point it at one of the archives to browse every section you wrote, panel by panel:
vibe-view open examples/vibe_view/runs/qvf_showcase/spectroscopy.qvf
The startup banner lists every section as rendered / skipped, and the
browser opens at http://127.0.0.1:8080. Walk the sidebar, each kind
this tutorial wrote has its own panel.
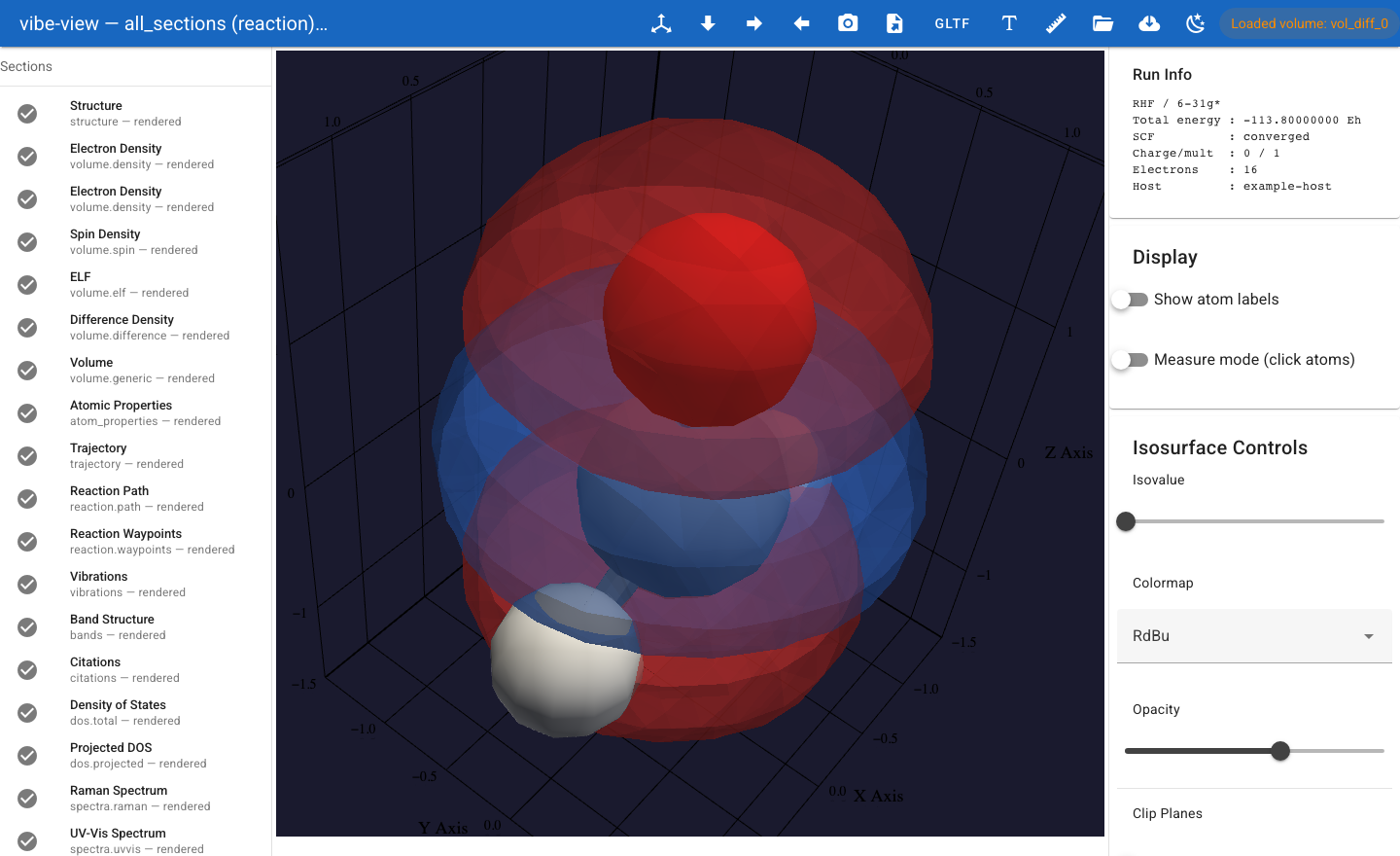
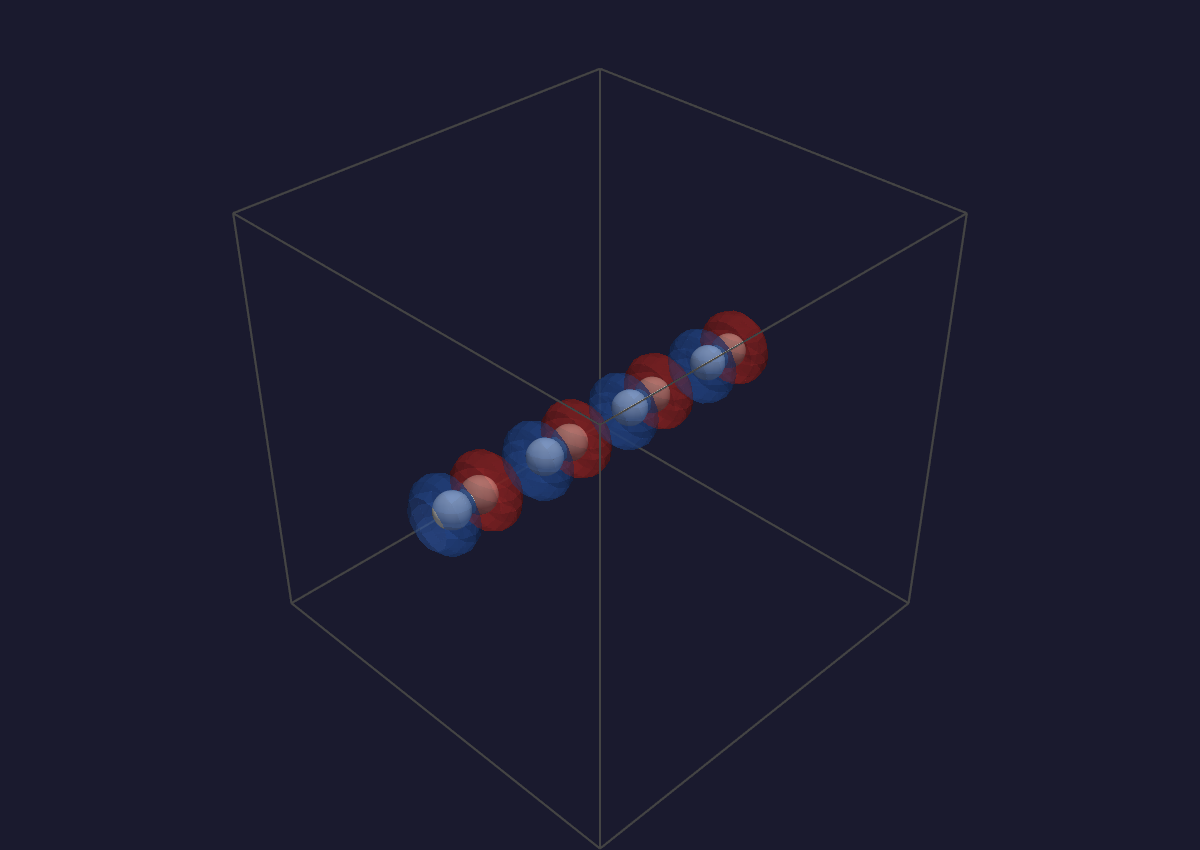
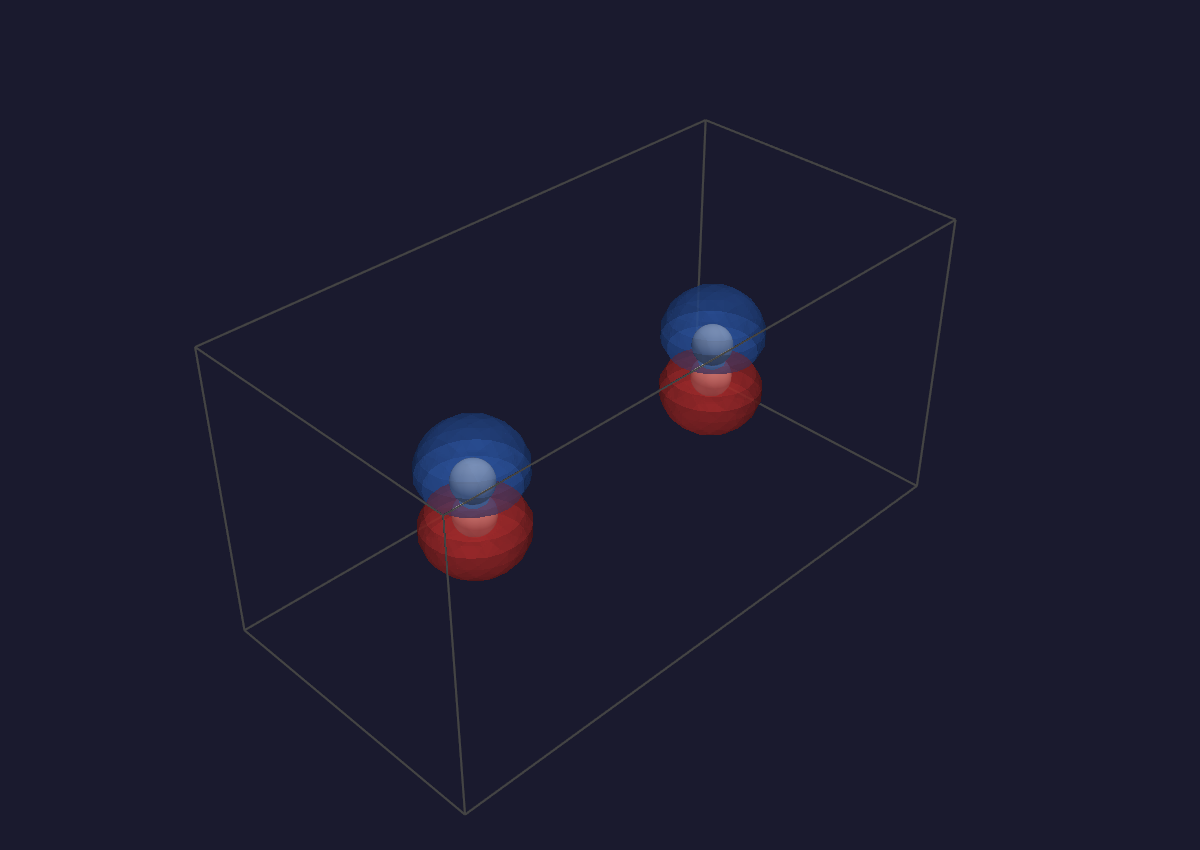
A two-colour difference-density isosurface (blue = positive, red =
negative) — one of the panels spectroscopy.qvf opens with.¶
See vibe-view: an end-to-end walkthrough for the panel-by-panel tour and the vibe-view user guide for every control.
8. Versioning, reserved kinds, and extensions¶
v1 vs v2. Current producers emit
qvf_version=1. Periodicreaction.pathandtrajectorysections may carry optional per-frame lattice vectors and dimensionality metadata, but those are additive v1 fields. The earlierqvf_version=2bump for periodic paths was withdrawn: consumers should still accept old archives labelledqvf_version=2as v1-compatible, but new producers should not emit that label.Reserved kinds. Portable canonical kinds live in the design-doc registry. If you need experimental data before a kind is promoted there, use
volume.generic,spectra.generic, or a vendor namespace such asx_<vendor>.*instead of inventing an unregistered canonical name.Vendor extensions. A producer can write its own section under the
x_<vendor>.*namespace. Consumers that do not understand it skip it (“skipped, vendor namespace”) rather than refusing the file, unless the section is flaggedcritical. The extension model is the governance for promoting a vendor kind to a canonical one.
Next¶
vibe-view walkthrough, the GUI tour of these sections.
QVF design doc, the full format specification: units, every section schema, the validation and extension models.
QVF consumer reference, read
.qvffiles programmatically without the viewer.auto citations, how the
citationssection is assembled.NEB reaction paths, the natural source of
reaction.pathsections.