Orbital and density visualization¶
Converged molecular orbitals and electron densities become useful when
you can see them. vibe-qc’s own visualization format is QVF
(.qvf, Quantum Visualization Format): one self-contained archive
carrying the structure, the density, the orbitals, and the basis + MO
coefficients together, opened
in vibe-view, vibe-qc’s GPU-accelerated viewer. That is the format
this tutorial leads with.
vibe-qc also writes the two universal interchange formats, for when a third-party viewer or a publication-grade render is what you need:
Molden (
.molden): one text file carrying geometry + basis + every MO. Jmol, Avogadro, IQmol, MolView all open it.Gaussian cube (
.cube): volumetric data on a 3D grid for isosurface rendering in VMD, PyMOL, ChimeraX, VESTA, ParaView, Blender.
Reach for QVF first: everything lands in one file, orbitals render with their sign, and the viewer can resample any MO on demand. Drop to Molden or cube when a specific external tool needs it.
The native format: .qvf for vibe-view¶
A single run_job call writes a complete .qvf archive: ask for
the density and orbitals you want, and turn on QVF output.
from vibeqc import Atom, Molecule, run_job
mol = Molecule([
Atom(8, [ 0.0, 0.00, 0.00]),
Atom(1, [ 0.0, 1.43, -0.98]),
Atom(1, [ 0.0, -1.43, -0.98]),
])
run_job(
mol, basis="6-31g*", method="rhf",
write_cube=["density", "homo", "lumo"], # which fields to evaluate
output_qvf=True, # bundle them into one .qvf
output="water",
)
# -> water.qvf
water.qvf is a single ZIP archive holding, as separate sections:
structure: the geometry;volume.density: the total electron density on a grid;volume.orbital: one section per requested MO (here HOMO and LUMO), each tagged with its energy and occupation;wavefunction.gto: the basis shells + MO coefficients, embedded becausewrite_molden_filedefaults toTrue, so the viewer can resample any orbital on demand without re-running vibe-qc.
Open it in vibe-view:
vibe-view open water.qvf
vibe-view renders the density as an isosurface and each orbital with
both signed lobes, and picks up every volume.orbital section in the
archive automatically.
Two dedicated tutorials cover this end to end: vibe-view: an end-to-end walkthrough is a full vibe-view walkthrough, and The QVF file format, end to end documents the QVF format section by section. The vibe-view guide has install instructions.
The same call also drops the individual water.density.cube,
water.homo.cube, and water.lumo.cube files next to the archive,
so the interchange cubes come for free.
Molden, for external molecular viewers¶
Every run_job call already writes a Molden file by default
(write_molden_file=True):
from vibeqc import Atom, Molecule, run_job
mol = Molecule([
Atom(8, [ 0.0, 0.00, 0.00]),
Atom(1, [ 0.0, 1.43, -0.98]),
Atom(1, [ 0.0, -1.43, -0.98]),
])
run_job(mol, basis="6-31g*", method="rhf", output="water")
# -> water.molden
Open it:
open -a Avogadro2 water.molden # macOS
# or
jmol water.molden # cross-platform
Select an MO from the energy list, render as an isosurface. For a publication-style plot you’ll usually want the cube route.
Cube files for single orbitals¶
QVF already embeds the density and orbital grids (above). Write
standalone .cube files when you want an external renderer (VMD,
PyMOL, …) or fine control over a single field:
import vibeqc as vq
mol = vq.Molecule([
vq.Atom(8, [ 0.0, 0.00, 0.00]),
vq.Atom(1, [ 0.0, 1.43, -0.98]),
vq.Atom(1, [ 0.0, -1.43, -0.98]),
])
basis = vq.BasisSet(mol, "6-31g*")
result = vq.run_rhf(mol, basis)
# HOMO is the highest doubly-occupied orbital (0-based index)
homo = mol.n_electrons() // 2 - 1
vq.write_cube_mo("water_homo.cube", result.mo_coeffs, homo, basis, mol)
vq.write_cube_mo("water_lumo.cube", result.mo_coeffs, homo + 1, basis, mol)
# Or write the full density
vq.write_cube_density("water_rho.cube", result.density, basis, mol)
The cube writer auto-picks a cubic bounding box and grid spacing appropriate for the molecule. For custom control see the volumetric data user guide.
Multiple MOs in one file¶
Some viewers (VMD, Avogadro) support multi-volume cube files, one file, several MOs, toggle between them in the UI.
homo = mol.n_electrons() // 2 - 1
vq.write_cube_mos(
"water_frontier.cube",
result.mo_coeffs,
orbital_indices=[homo - 1, homo, homo + 1, homo + 2],
basis=basis,
molecule=mol,
)
Single, double, and triple bonds¶
A single bond is a single σ orbital; a double bond is σ plus one π; a triple bond is σ plus two perpendicular π. The σ is cylindrically symmetric about the bond axis, while each π has a nodal plane through the axis and lobes above and below it, so the π orbitals are the visible signature of multiple-bond character in an MO plot.
The π bonds sit at or near the HOMO, which makes them easy to pull out. Ethylene’s C=C π is the HOMO; acetylene’s C≡C carries a degenerate pair of π orbitals as its HOMO and HOMO-1:
import vibeqc as vq
# Double bond: ethylene C2H4 (planar, in the xy-plane). HOMO = π(C=C).
ethylene = vq.Molecule([
vq.Atom(6, [ 1.265, 0.000, 0.0]),
vq.Atom(6, [-1.265, 0.000, 0.0]),
vq.Atom(1, [ 2.340, 1.740, 0.0]),
vq.Atom(1, [ 2.340, -1.740, 0.0]),
vq.Atom(1, [-2.340, 1.740, 0.0]),
vq.Atom(1, [-2.340, -1.740, 0.0]),
])
vq.run_job(ethylene, basis="6-31g*", method="rhf",
write_cube=["homo"], output_qvf=True, output="ethylene")
# ethylene.qvf -> vibe-view: the HOMO is the C=C π, two lobes above and
# below the molecular plane.
# Triple bond: acetylene C2H2 (linear along z). HOMO + HOMO-1 = the two π.
acetylene = vq.Molecule([
vq.Atom(6, [0.0, 0.0, 1.136]),
vq.Atom(6, [0.0, 0.0, -1.136]),
vq.Atom(1, [0.0, 0.0, 3.141]),
vq.Atom(1, [0.0, 0.0, -3.141]),
])
vq.run_job(acetylene, basis="6-31g*", method="rhf",
write_cube=["homo", "homo-1"], output_qvf=True, output="acetylene")
# acetylene.qvf -> vibe-view: HOMO and HOMO-1 are the two perpendicular
# π bonds of the C≡C triple bond; the σ(C-C) sits just below them.
Open each .qvf in vibe-view and step through the orbitals: a single
bond gives only the cylindrical σ, ethylene adds one π, and acetylene
two. The σ alone is what a single bond looks like; the perpendicular π
lobes are what set a double or triple bond apart.
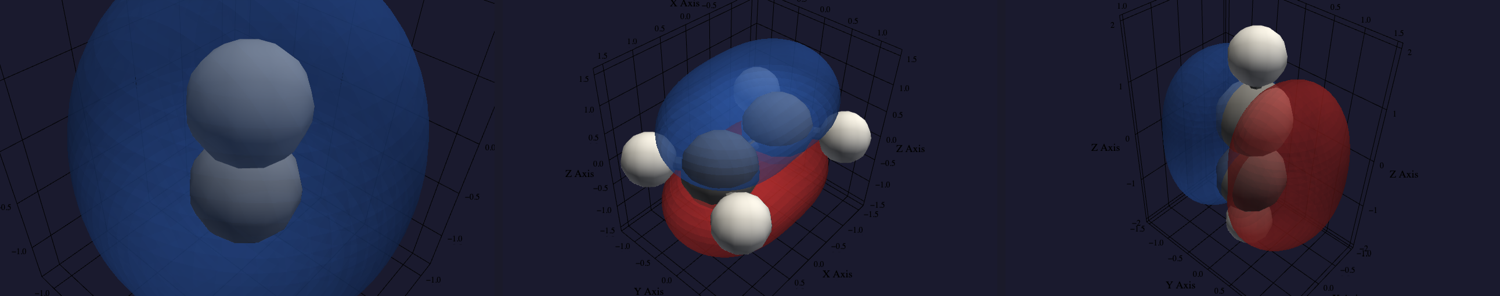
The bonding orbitals rendered in vibe-view (signed lobes: blue positive,
red negative). Left: H₂, the σ single bond, one lobe of a single sign.
Centre: ethylene, the C=C π that makes the double bond, with lobes above
and below the molecular plane. Right: acetylene, one of the two
perpendicular C≡C π orbitals of the triple bond. Regenerated by
[examples/plots/bond-orbitals.py](https://gitlab.peintinger.com/mpei/vibeqc/-/blob/main/examples/plots/bond-orbitals.py).¶
Viewer recipes¶
Given water_homo.cube on disk:
VMD (publication-quality rendering with Tachyon/POV-Ray):
vmd water_homo.cube
# In the TkCon:
mol rename top water
mol addrep top
mol modstyle 1 top Isosurface 0.05 0 0 0 1 1
Avogadro 2, open the cube, View → Add View → Orbitals, pick the isovalue (0.03-0.05 is a good starting point for MOs).
PyMOL:
load water_homo.cube
isomesh homo_pos, water_homo, 0.05
isomesh homo_neg, water_homo, -0.05
color cyan, homo_pos
color orange, homo_neg
Blender (with Molecular Nodes): directly imports cube as a volumetric object, isosurface is a geometry-nodes tree away.
Grid settings¶
The default grid (0.2 bohr spacing, 3 bohr padding around atoms) is fine for visualization. For tighter or sparser meshes:
vq.write_cube_mo(
"water_homo_fine.cube",
result.mo_coeffs, homo, basis, mol,
spacing=0.1, # bohr
padding=5.0, # bohr around the bounding box of atoms
)
A finer grid gives cleaner isosurface curvature at the cost of file
size; 0.1 bohr is typical for papers, 0.2 bohr is plenty for quick
looks. The file size scales as 1 / spacing³.
Inline preview: 2D contour cuts¶
If you don’t have an external viewer at hand, vibe-qc’s
evaluate_ao lets you sample any MO or the density on an arbitrary
set of points and render 2D slices in pure matplotlib. This is the
fast way to check an orbital before launching VMD / Avogadro for the
publication-quality 3D rendering.
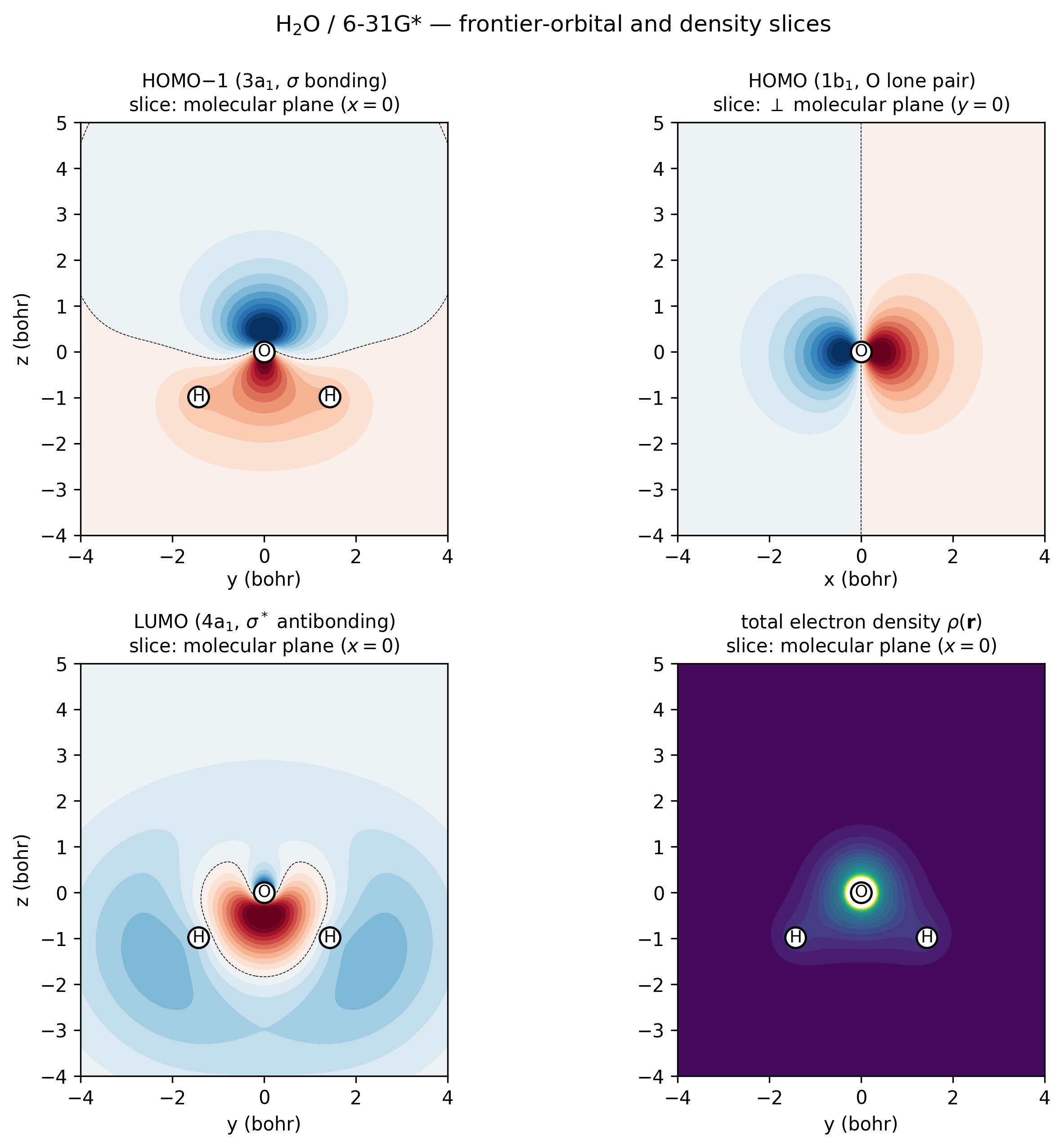
H₂O / 6-31G* frontier orbitals and density, sampled directly via
vibeqc.evaluate_ao and rendered as 2D contour slices.
Top-left (3a₁ HOMO−1, \(\sigma\)-bonding): in-phase combination of
the two O–H bonds with positive amplitude all along both bonds —
the “valence σ” of water. Top-right (1b₁ HOMO, oxygen lone
pair): the orbital is perpendicular to the molecular plane, so
this slice goes through O at \(y = 0\) and shows the textbook
two-lobe ±x dumbbell. Bottom-left (4a₁ LUMO,
\(\sigma^*\)-antibonding): nodal plane between O and the H–H
midpoint, lobes pointing away from H atoms. Bottom-right
(total density): cusps at every nucleus and the smooth bonding
ridge between O and each H. Regenerated by
[examples/plots/water-frontier-orbitals.py](https://gitlab.peintinger.com/mpei/vibeqc/-/blob/main/examples/plots/water-frontier-orbitals.py).¶
The 2D-slice route uses the same AO evaluator that write_cube_*
uses internally, so the numbers are bit-identical to a thin slice
through the cube file, but you skip the disk-write / external-tool
round-trip when all you want is a sanity check.
Caveats¶
HF orbitals aren’t “truth”. Molecular orbitals are a representation-dependent quantity, Kohn-Sham orbitals from the same basis set often look visibly different. When comparing to the literature, note the method that generated them.
Phase arbitrariness. The overall sign of an MO is arbitrary, two SCF runs can land on C_i → +C_i or −C_i. The density is the physical quantity; the color of HOMO lobes can flip between runs.
Tighter basis → more structure. Minimal-basis HOMOs look bland; by TZVP you start seeing polarization lobes that are genuinely chemically informative.
Theory¶
The sections below explain what these pictures actually are: the math an orbital isosurface renders, the three file formats vibe-qc writes (QVF, cube, Molden) and how each stores the data, what a finer grid costs, and why an orbital’s overall sign is arbitrary.
What an “orbital isosurface” actually plots¶
A molecular orbital \(\phi_i(\mathbf{r}) = \sum_\mu C_{\mu i} \chi_\mu(\mathbf{r})\) is a real-valued function of \(\mathbf{r}\) in three-dimensional space (imaginary for k-space bands; see Band structure and density of states for those). What viewers call an “isosurface at value \(c\)” is the mathematical surface
rendered as a triangulated mesh. The marching-cubes algorithm (Lorensen-Cline 1987) converts the scalar field on a regular grid into this mesh; every mainstream viewer (VMD, PyMOL, Avogadro, ParaView) uses it or a close variant.
Common rendering conventions:
Color the two signs differently. Since \(\phi\) is real but signed, isosurfaces at \(+c\) and \(-c\) exist simultaneously. Showing both in complementary colors (cyan / orange, red / blue) makes the nodal structure visible.
Isovalue choice. \(c = 0.05\) bohr⁻³ᐟ² is a good default that captures the bonding lobes without drowning in the diffuse tail. Tighten to 0.08 for a compact view of valence MOs; loosen to 0.02 to see Rydberg-like tails.
The density \(\rho(\mathbf{r}) = \sum_i |\phi_i|^2\) is always positive and sign-ambiguous, an isosurface at \(\rho = 0.01\) shows electron localisation without the phase-flip artefact.
The QVF format¶
vibe-qc’s native format, QVF (.qvf), is a ZIP container with a
JSON manifest that holds one calculation’s data as typed sections: the
structure, the volume.density and volume.orbital grids this
tutorial produces, the wavefunction.gto basis + coefficients, and
many more (bands, spectra, trajectories, vibrational modes). Unlike
cube and Molden, it keeps everything for a calculation in a single
random-access file, and records each orbital’s energy, occupation, and
spin alongside its grid.
The format is open, versioned, and has a producer/consumer contract, so it is not a vibe-qc-only convention. Its full specification is the QVF design doc / tech spec (section kinds, units, extension and versioning model); the rendered docs also carry a hands-on The QVF file format, end to end that walks the format end to end and vibe-view: an end-to-end walkthrough that drives the viewer. The motivation behind the format is laid out in the blog post Quantum chemistry needs a modern file format.
The Gaussian cube format¶
The cube writer samples the scalar field on a regular Cartesian grid and writes a plain-ASCII file. The structure:
Comment line 1
Comment line 2
n_atoms x0 y0 z0 <- origin of the grid (bohr)
n_x dx 0 0 <- grid vectors (bohr), direction and spacing
n_y 0 dy 0
n_z 0 0 dz
Z_1 Q_1 x_1 y_1 z_1 <- atoms: atomic number, nuclear charge, position
...
<floating-point values, in scanline order>
vibe-qc’s write_cube_mo evaluates \(\phi_i(\mathbf{r}_g)\) at
every grid point \(\mathbf{r}_g\) by direct AO evaluation on the grid
(uses the same evaluate_ao machinery that the DFT XC grid does
internally) followed by the AO-to-MO contraction
\(\phi_i(\mathbf{r}_g) = \sum_\mu C_{\mu i} \chi_\mu(\mathbf{r}_g)\).
The write_cube_density path contracts against the density matrix
instead: \(\rho(\mathbf{r}_g) = \sum_{\mu\nu} P_{\mu\nu} \chi_\mu(\mathbf{r}_g)
\chi_\nu(\mathbf{r}_g)\).
The Molden format¶
A Molden file is a single plain-text file that carries the whole
electronic structure rather than one sampled field. vibe-qc’s writer
emits a [Molden Format] header followed by:
[Atoms]: the geometry (in atomic units);[GTO]: the basis, as per-atom Gaussian shell listings;a spherical-harmonic flag (
[5D]/[7F]/[9G]) when the basis is solid-harmonic;[MO]: every molecular orbital as a list of AO coefficients, each tagged with its energy (Ene=), spin (Spin=), and occupation (Occup=). An unrestricted (UHF / UKS) result writes two[MO]blocks, alpha then beta.
Because it stores the basis and the coefficients instead of a
pre-sampled grid, a Molden viewer resamples each orbital on its own
grid at whatever isovalue you pick. That is the same on-demand
resampling the wavefunction.gto QVF section gives vibe-view: no grid
to choose and a small file, at the cost of depending on the viewer’s
AO evaluator rather than vibe-qc’s.
Grid cost¶
File size scales as \(1 / \text{spacing}^3\). At 0.2 bohr on a water molecule with 3 bohr padding, that’s \(\sim 40^3 \approx 6 \times 10^4\) values; at 0.1 bohr, \(\sim 5 \times 10^5\). AO evaluation cost scales linearly in grid points and quadratically in basis functions, so doubling the resolution makes the file 8× bigger and the write 8× slower. Use 0.2 for quick looks, 0.1 for papers.
The sign-arbitrariness caveat¶
A single-reference SCF converges to a Slater determinant whose columns \(\mathbf{C}_i\) are eigenvectors of \(\mathbf{F}\). Eigenvectors are defined only up to a sign (real) or phase (complex), so the overall sign of an MO plotted on different SCF runs can flip. The density is invariant, \(|\phi_i|^2\) is well-defined, but the colored lobes of a rendered MO can swap between runs. When comparing MO pictures across codes, or across functionals, don’t chase the lobe-color pattern; chase the nodal structure.
Resources¶
~250 MB peak RAM, ~3 s on one core (Apple M2 baseline) for the H₂O / 6-31G* HF + cube-write at 0.2 bohr spacing. Cube-file size scales as \(1/\text{spacing}^3\); the AO evaluation on the grid scales linearly in grid points and quadratically in basis size, so dropping to 0.1 bohr makes the file 8× bigger and the write 8× slower.
References¶
Marching cubes. W. E. Lorensen and H. E. Cline, “Marching cubes: a high resolution 3D surface construction algorithm,” ACM SIGGRAPH Comput. Graph. 21, 163 (1987). The basis of every isosurface renderer in chemistry.
Gaussian cube file format. Defined informally in the Gaussian programme’s documentation; community-standardised description at https://paulbourke.net/dataformats/cube/ and the
cubegensection of the Gaussian manual. vibe-qc’s writer matches the conventions Avogadro / VMD / PyMOL all consume.Textbook treatment of MO visualization. W. J. Hehre, L. Radom, P. V. R. Schleyer, and J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley (1986), foundational MO pictures of organic molecules that set the conventions still in use.
Next¶
Periodic systems get a .qvf too: run_periodic_job(..., output_qvf=True) bundles the structure, density, and (where computed)
bands and DOS into one archive that vibe-view renders the same way. See
the vibe-view guide.
For the plain-text periodic volumetric formats, XSF files play the role
cube files do for molecules: see the
volumetric-data user guide for the
equivalents (write_xsf_volume, write_bxsf for Fermi surfaces).